REDD1 loss reprograms lipid metabolism to drive progression of RAS mutant tumors
Publication:
Genes Dev 2020 Jun 1;34(11-12):751-766.
Authors:
Shuxi Qiao # 1 2, Siang-Boon Koh # 1 2, Varunika Vivekanandan 1, Devika Salunke 1, Krushna Chandra Patra 1 2, Elma Zaganjor 3, Kenneth Ross 1 2 4, Yusuke Mizukami 1 2, Sarah Jeanfavre 4, Athena Chen 2 5, Mari Mino-Kenudson 1 2 5, Sridhar Ramaswamy 1 2 3 4, Clary Clish 4, Marcia Haigis 3, Nabeel Bardeesy 1 2, Leif W Ellisen 1
Abstract:
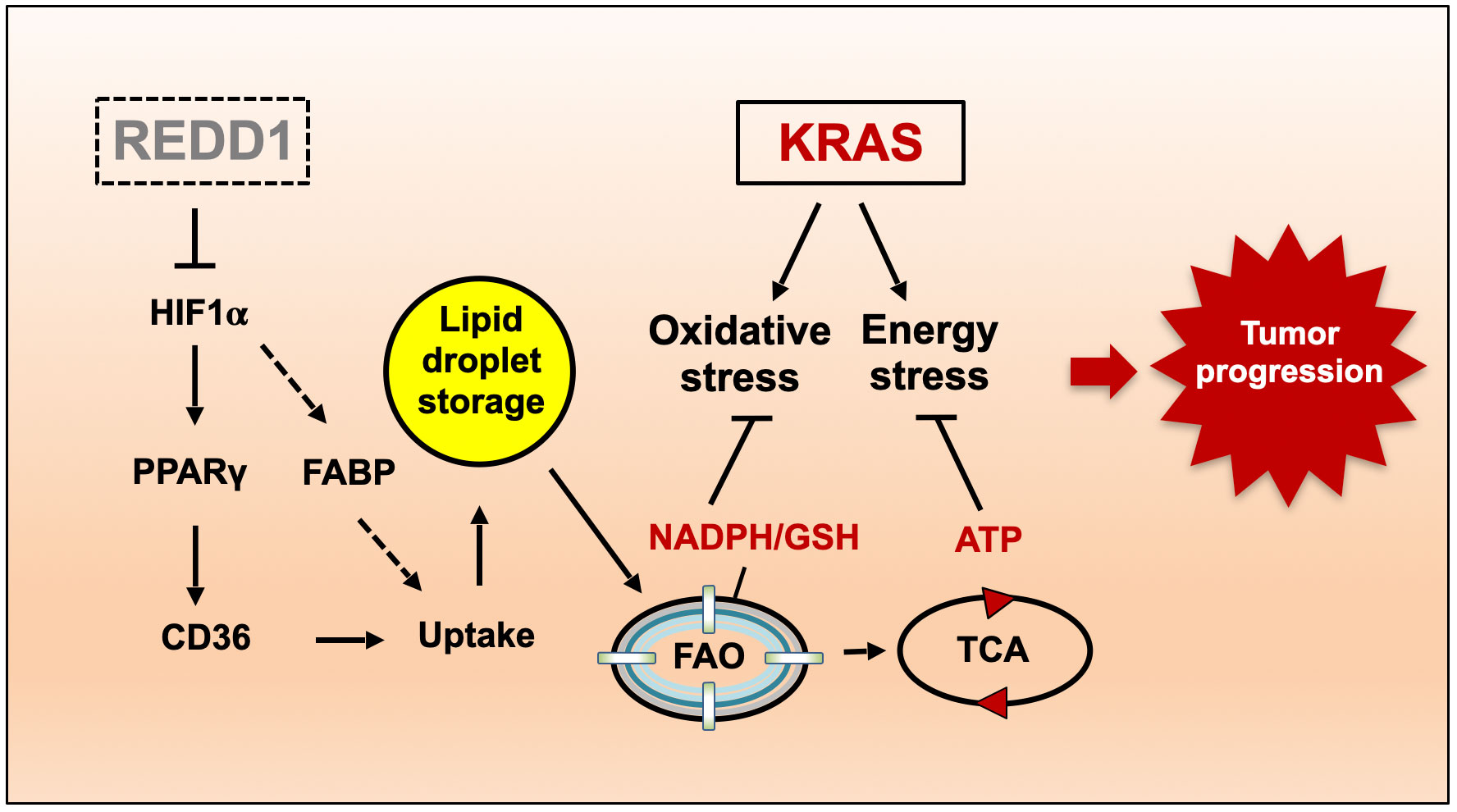
Human cancers with activating RAS mutations are typically highly aggressive and treatment-refractory, yet RAS mutation itself is insufficient for tumorigenesis, due in part to profound metabolic stress induced by RAS activation. Here we show that loss of REDD1, a stress-induced metabolic regulator, is sufficient to reprogram lipid metabolism and drive progression of RAS mutant cancers. Redd1 deletion in genetically engineered mouse models (GEMMs) of KRAS-dependent pancreatic and lung adenocarcinomas converts preneoplastic lesions into invasive and metastatic carcinomas. Metabolic profiling reveals that REDD1-deficient/RAS mutant cells exhibit enhanced uptake of lysophospholipids and lipid storage, coupled to augmented fatty acid oxidation that sustains both ATP levels and ROS-detoxifying NADPH. Mechanistically, REDD1 loss triggers HIF-dependent activation of a lipid storage pathway involving PPARγ and the prometastatic factor CD36. Correspondingly, decreased REDD1 expression and a signature of REDD1 loss predict poor outcomes selectively in RAS mutant but not RAS wild-type human lung and pancreas carcinomas. Collectively, our findings reveal the REDD1-mediated stress response as a novel tumor suppressor whose loss defines a RAS mutant tumor subset characterized by reprogramming of lipid metabolism, invasive and metastatic progression, and poor prognosis. This work thus provides new mechanistic and clinically relevant insights into the phenotypic heterogeneity and metabolic rewiring that underlies these common cancers.