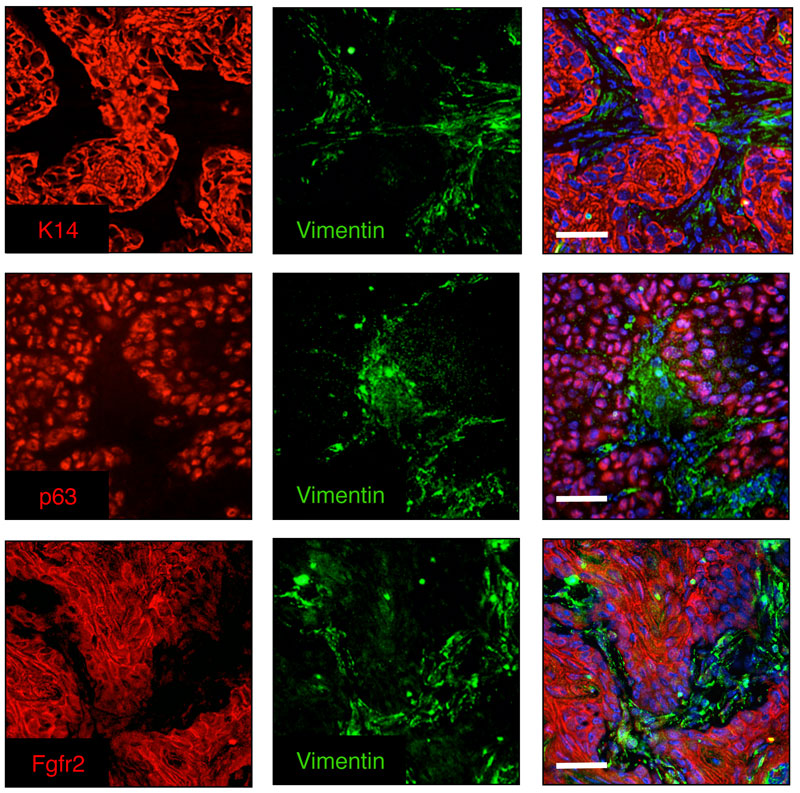
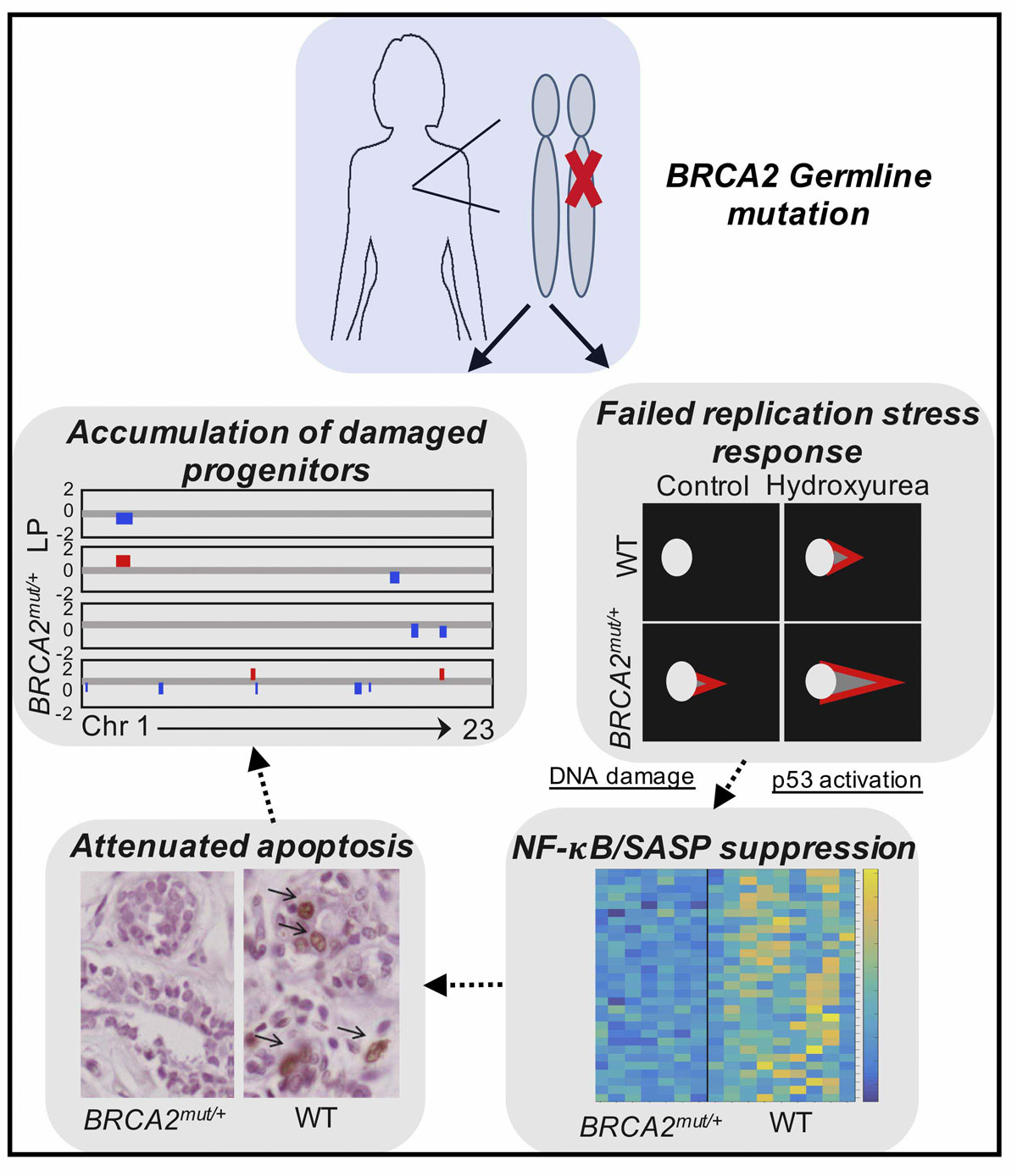
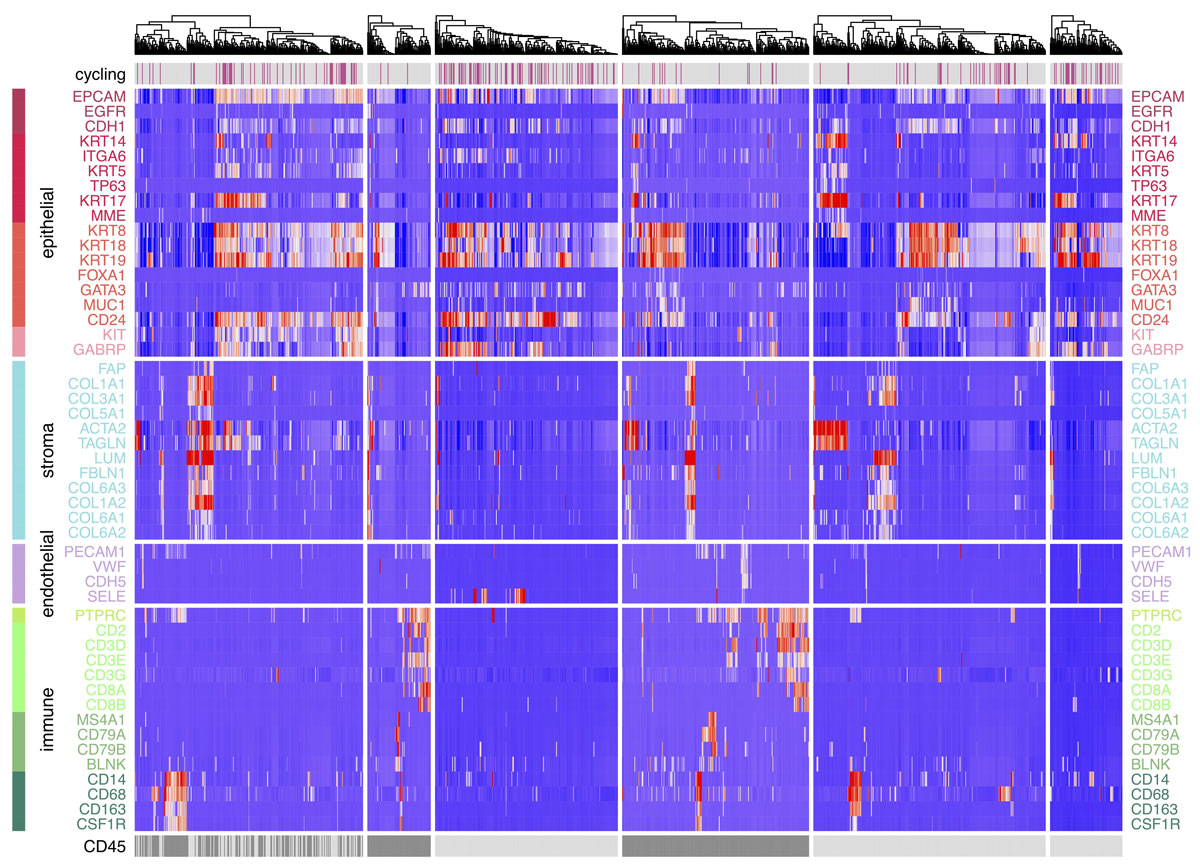
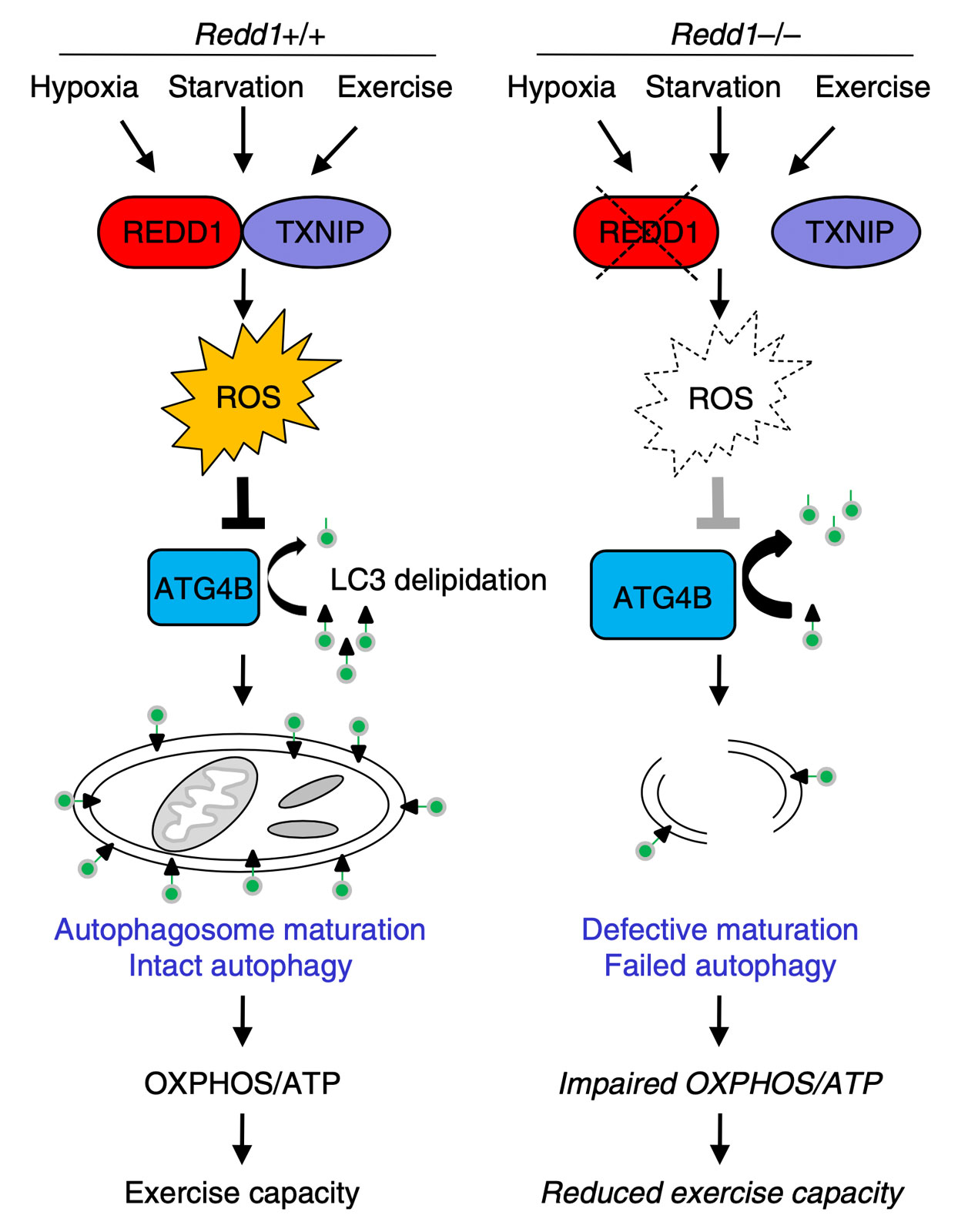
Our group is broadly interested in how genetic abnormalities in breast cancer and related malignancies influence tumor biology, and how that biology can, in turn, be exploited to therapeutic advantage.
We address these questions through basic research studies of key cancer drivers including DNA repair defects through BRCA1/2 and related pathways, and transcriptional reprogramming through the p53 gene family. Supporting and complementing these studies are leading-edge analyses of patient-derived precancerous and cancerous tissues. Recent innovative tissue-based studies have led to our discovery of novel cancer drivers, and have provided a unique window on early cancer pathogenesis, intratumoral heterogeneity and tumor progression. Our discoveries in the basic laboratory and through human tumor analysis are being applied in ongoing clinical trials that seek to identify predictive markers of response to specific therapeutics for breast and other cancers. Our ability to work at the interface of basic tumor biology and therapeutic application is strongly supported by our network of collaborators and by the research and clinical infrastructure of the Mass General Cancer Center.